Molecular Projected DOS
NO molecule on Ni(001)
For this example we calculate the projected MOs of a NO molecule on a Ni(001) slab. In the following the required input files are:
no-on-ni001.param, no-on-ni001.cell, no-on-ni001.molpdos, gasphase.cell, gasphase.param, gasphase.check
no-on-ni001.param
calculate_modos : true
deltascf_checkpoint : gasphase
task: SinglePoint
spin_polarized : True
cut_off_energy : 400.0
elec_energy_tol : 1e-07
fix_occupancy : False
iprint : 1
max_scf_cycles : 200
metals_method : dm
mixing_scheme : Pulay
nextra_bands : 10
num_dump_cycles : 0
opt_strategy_bias : 3
smearing_scheme : Gaussian
smearing_width : 0.1
xc_functional : RPBE
no-on-ni001.cell
%BLOCK LATTICE_CART
3.5240000000 0.0000000000 0.0000000000
0.0000000000 3.5240000000 0.0000000000
0.0000000000 0.0000000000 23.0000000000
%ENDBLOCK LATTICE_CART
%BLOCK POSITIONS_ABS
Ni 1.762000 0.000000 1.762000
Ni 0.000000 1.762000 1.762000
Ni 0.000000 0.000000 3.524000
Ni 1.762000 1.762000 3.524000
Ni 1.762000 0.000000 5.286000
Ni 0.000000 1.762000 5.286000
N 1.7620 0.0000 7.0196
O 1.7620 -0.0000 8.1902
%ENDBLOCK POSITIONS_ABS
%BLOCK IONIC_CONSTRAINTS
1 Ni 1 1 0 0
2 Ni 1 0 1 0
3 Ni 1 0 0 1
4 Ni 2 1 0 0
5 Ni 2 0 1 0
6 Ni 2 0 0 1
7 Ni 3 1 0 0
8 Ni 3 0 1 0
9 Ni 3 0 0 1
10 Ni 4 1 0 0
11 Ni 4 0 1 0
12 Ni 4 0 0 1
13 Ni 5 1 0 0
14 Ni 5 0 1 0
15 Ni 5 0 0 1
16 Ni 6 1 0 0
17 Ni 6 0 1 0
18 Ni 6 0 0 1
%ENDBLOCK IONIC_CONSTRAINTS
FIX_ALL_CELL : True
KPOINTS_MP_GRID : 2 2 1
KPOINTS_MP_OFFSET : 0.25 0.25 0.25
no-on-ni001.molpdos
molpdos_state : 4 1
molpdos_state : 5 1
molpdos_state : 6 1
molpdos_state : 4 2
molpdos_state : 5 2
molpdos_state : 6 2
molpdos_bin_width : 0.01
molpdos_smearing : 0.10
molpdos_scaling : 1.00
axis_energy_margin : 2.00
output_filename : MolPDOS.dat
gasphase.cell
%BLOCK LATTICE_CART
3.5240000000 0.0000000000 0.0000000000
0.0000000000 3.5240000000 0.0000000000
0.0000000000 0.0000000000 23.0000000000
%ENDBLOCK LATTICE_CART
%BLOCK POSITIONS_ABS
N 1.7620 0.0000 7.0196
O 1.7620 -0.0000 8.1902
%ENDBLOCK POSITIONS_ABS
FIX_ALL_CELL : True
KPOINTS_MP_GRID : 2 2 1
KPOINTS_MP_OFFSET : 0.25 0.25 0.25
gasphase.param
task: SinglePoint
spin_polarized : True
cut_off_energy : 400.0
elec_energy_tol : 1e-07
fix_occupancy : False
iprint : 1
max_scf_cycles : 200
metals_method : dm
mixing_scheme : Pulay
nextra_bands : 10
num_dump_cycles : 0
opt_strategy_bias : 3
smearing_scheme : Gaussian
smearing_width : 0.1
xc_functional : RPBE
After generating gasphase.check by running CASTEP on the gasphase.param and gasphase.cell files, we execute CASTEP and post-process with MolPDOS. This will write x-y data files for the Total DOS, the separate spin channels, and the MolPDOS peaks.
The following image shows the Total DOS and the two spin channels.
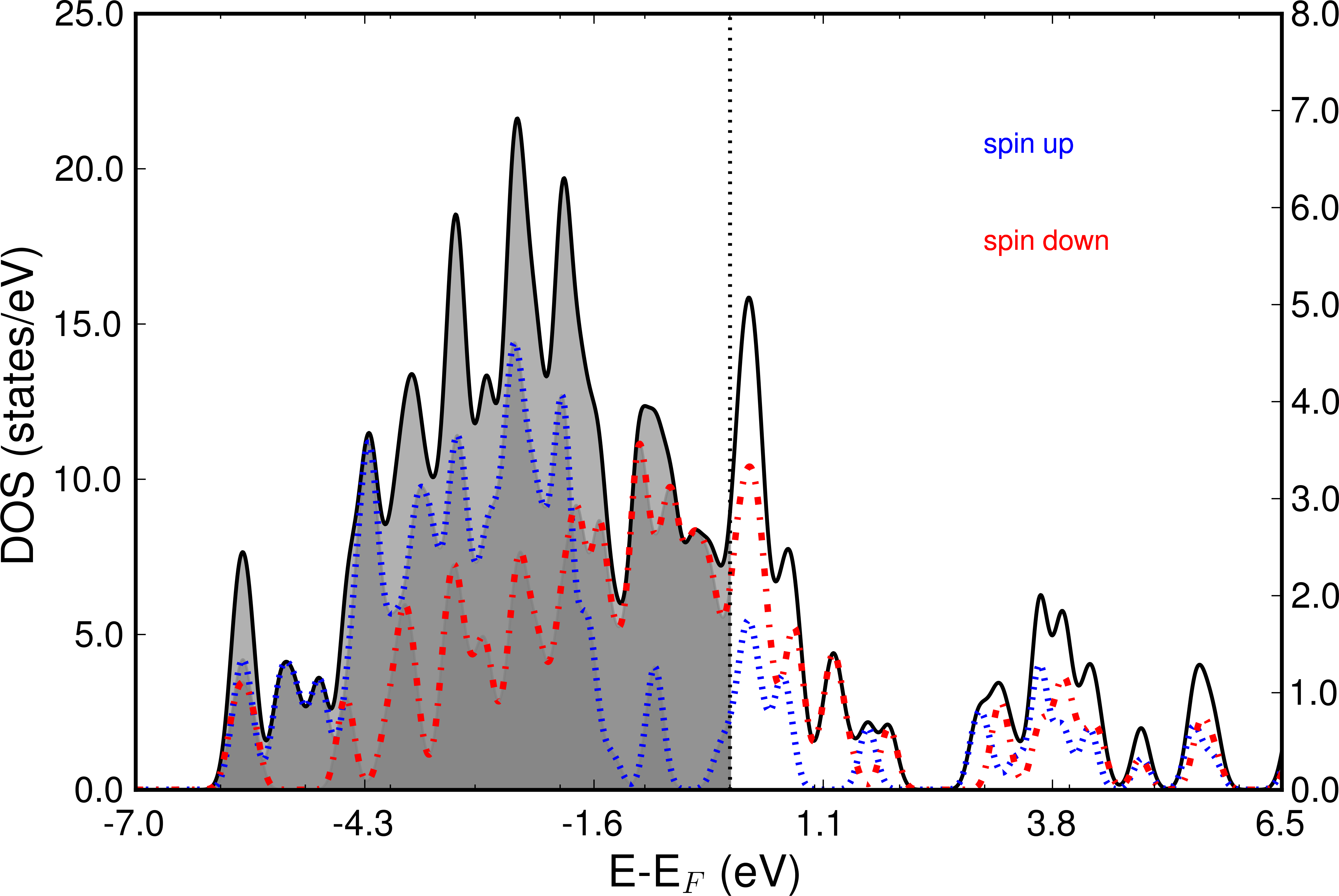
The next picture shows the frontier orbitals of spin channel 1 projected on the total DOS. Especially the LUMO shows strong hybridization with the Nickel d-bands and also is partially occupied. The left scale refers to the total DOS, whereas the right y-scale shows the peak height of the projected MOs.
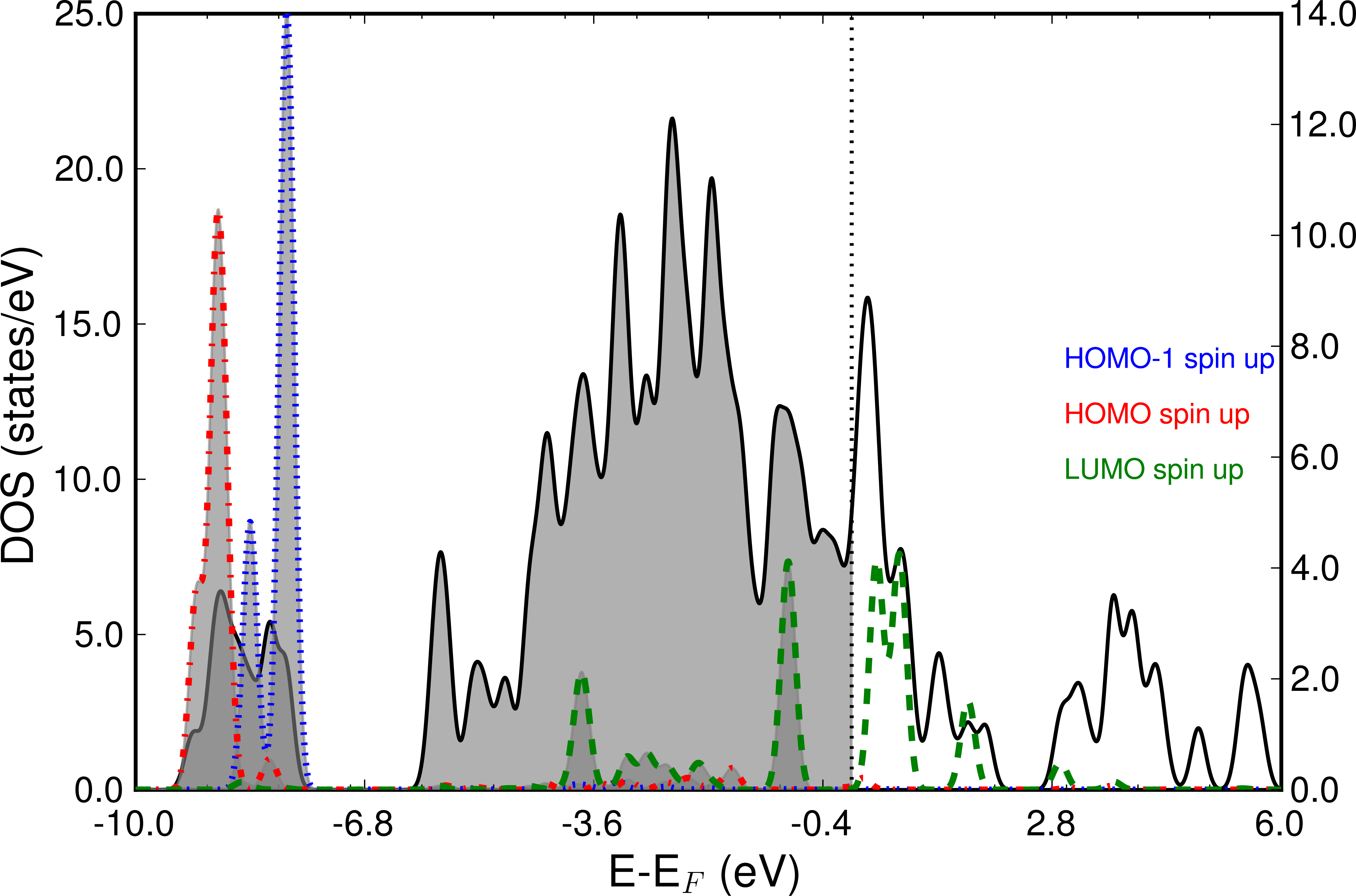
GOOD TO KNOW
If you ever forget the correct input for <seed>.molpdos, just run the MolPDOS tool without seed. The printed information is all you need!